Contributo di Martino Bolognesi (Università degli Studi di Milano) per SIB
Recentemente, le applicazioni dell’Intelligenza Artificiale (IA) hanno trovato ampio utilizzo in vari settori, inclusi quelli personali, sociali, economici e militari. Anche la ricerca scientifica, in particolare le scienze biomediche e della vita, ha ricevuto importanti contributi dall’IA. Ad esempio, la diagnostica medica per immagini è sensibilmente migliorata grazie all’applicazione di IA istruita dall’analisi di milioni di immagini e diagnosi passate.
Nel campo della biochimica, l’IA ha ottenuto risultati straordinari, risolvendo il problema della previsione della struttura tridimensionale (3D) delle proteine a partire dalla loro sequenza aminoacidica, una sfida scientifica aperta da decenni. Negli anni ’70 del secolo scorso, il Premio Nobel Christian Anfinsen dimostrò che la struttura 3D delle proteine poteva essere ripristinata reversibilmente, dopo averla sensibilmente perturbata; ciò suggeriva che la sequenza degli aminoacidi di una proteina contiene già tutte le informazioni necessarie per portarla alla sua corretta struttura 3D ripiegata.
Perché questa scoperta è importante?
Abbiamo due risposte:
1. Determinare la sequenza degli aminoacidi costituenti una proteina è un processo sperimentalmente accessibile con facilità (anche a partire dalle informazioni genomiche);
2. La struttura 3D di una proteina ne spiega i meccanismi di azione in vivo ed è la base per molte applicazioni pratiche, ad esempio per progettare farmaci potenti e specifici.
Si può comprendere, pertanto, quanto la sfida della previsione accurata delle strutture proteiche abbia interessato i ricercatori biochimici e biofisici da almeno 50 anni.
Nonostante un’ampia serie di idee e approcci basati su principi chimico-fisici e computazionali, fino al 2019 le previsioni prodotte erano solo in parte soddisfacenti, e soprattutto erano scarsamente performanti nella previsione di strutture 3D di proteine assenti dai data-base di riferimento.
Questo panorama ha registrato un passo avanti, che potremmo definire epocale, quando (2018) i metodi computazionali basati su reti neurali e deep learning sono stati impiegati per analizzare le strutture proteiche 3D note (circa 200.000) ed estrarre informazioni innovative, per esempio sui contatti interatomici e le distribuzioni di distanze nel cuore delle proteine globulari. Su queste basi, DeepMind (un’azienda di ricerca sussidiaria di GOOGLE), sotto la guida di Demis Hassabis, ha prodotto e distribuito AlphaFold (AF1), un approccio computazionale che ha stupito la comunità scientifica per l’accuratezza delle previsioni, decisamente migliori di quelle ottenute da tutti gli approcci precedenti. AF1 è stato paragonato a un super maestro di origami, che dalla sequenza lineare di aminoacidi produce (in qualche decina di minuti) una struttura 3D indistinguibile da quella determinata sperimentalmente (in mesi) della stessa proteina.
A fronte di questo spettacolare successo, Hassabis e il collega John Jumper hanno successivamente sviluppato versioni più avanzate, AF2 e AF3 (2020 e 2024), che si fondano su miglioramenti degli algoritmi e delle procedure di ‘learning’, e compiono un passo avanti nello studio della complessità. Infatti, mentre AF1 ha mietuto successi nella previsione delle strutture 3D di proteine monomeriche, AF2 e particolarmente AF3 si sono orientati verso la previsione della struttura di complessi macromolecolari, quali proteine oligomeriche, complessi tra proteine diverse, complessi di proteine con altre classi (macro)molecolari quali gli acidi nucleici. L’accuratezza di questi strumenti costituisce un enorme passo avanti, anche per le scienze applicate.
Un filone di ricerca distinto da quello della previsione della struttura 3D delle proteine riguarda il problema inverso: come produrre una proteina con una nuova e specifica struttura 3D attraverso la codifica della sua sequenza aminoacidica. Anche in questo caso, il progresso è stato guidato dalla crescente conoscenza di strutture proteiche, dei principi chimico-fisici che le caratterizzano, e dall’applicazione di metodi di IA. Il gruppo di David Baker, fin dai primi anni 2000, ha coltivato questo campo con approcci progressivamente più avanzati (codificati nel pacchetto di software ROSETTA) dedicandosi alla progettazione di proteine con funzioni specifiche, quali attività enzimatiche o proprietà di legame per steroidi o farmaci, ma anche per produrre nuovi nanomateriali a base proteica.
In conclusione, AlphaFold e ROSETTA rappresentano una rivoluzione scientifica attesa da lungo tempo. L’IA ha accelerato queste scoperte, fornendo nuovi stimoli per lo studio dei meccanismi cellulari, delle patologie e dell’azione dei farmaci, fornendo importanti stimoli e input per le biotecnologie. In riconoscimento di questi progressi, la Royal Swedish Academy of Sciences ha assegnato il Premio Nobel per la Chimica 2024 a David Baker, Demis Hassabis e John Jumper “per la progettazione computazionale delle proteine e la previsione delle loro strutture”.
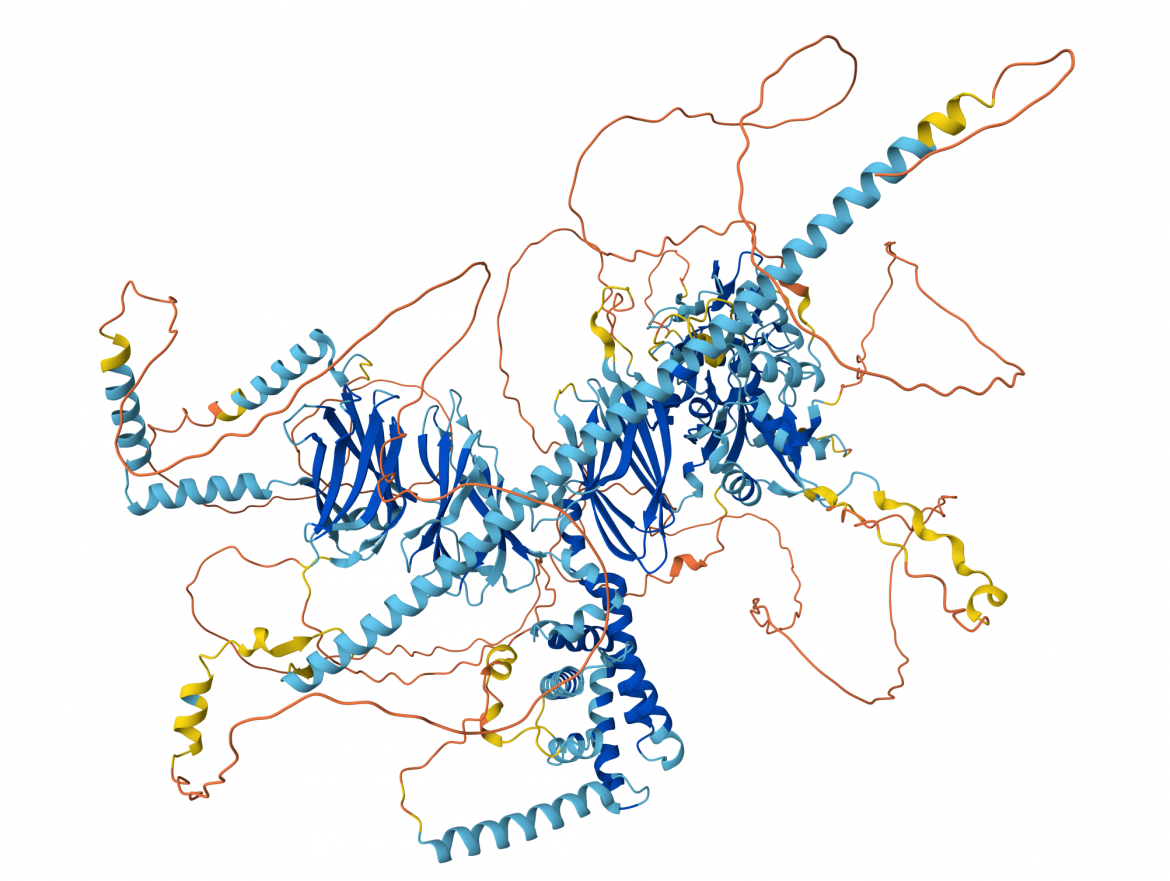
* IN FOTO: Previsione della struttura 3D della Androglobina, una proteina particolarmente complessa, composta da 1667 aminoacidi organizzati in più domini strutturali e funzionali. L’attività di questa proteina è essenziale per la corretta formazione e maturazione degli spermatozoi e svolge un ruolo centrale nel controllo della fertilità maschile. L’attendibilità della previsione strutturale è codificata dai colori che vanno dal blu (alta attendibilità) al rosso (bassa); i segmenti rappresentati in tratto sottile rappresentano zone della proteina a struttura non prevedibile perché probabilmente intrinsecamente disordinate. Immagine tratta dal AlphaFold Protein Structure Database